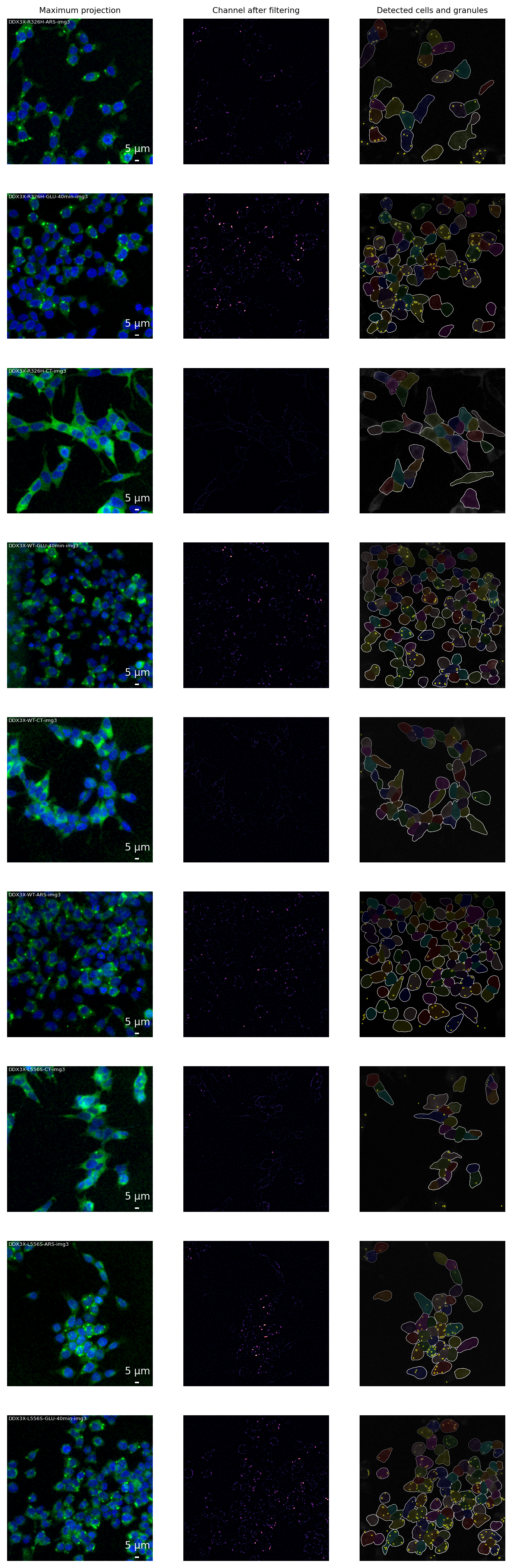
Confocal microscopy images were analyzed using a custom Python pipeline. Maximum intensity Z-projections were generated for both DAPI and GFP channels. For granule detection, the GFP channel was preprocessed using Difference of Gaussians filtering (σ₁ = 3.0, σ₂ = 4.8) to enhance punctate structures while suppressing background noise. Granules were segmented using a global intensity threshold of 0.0009, followed by removal of objects smaller than 15 pixels and those touching image borders.
Cell segmentation was performed using Cellpose v3.1 (Stringer et al. 2021) with the cyto3 model (Stringer and Pachitariu 2024). Prior to segmentation, images were preprocessed with Gaussian smoothing (σ = 0.5 for GFP and σ = 1.0 for DAPI channels) and intensity rescaling to the [0,1] range. The model was configured with a target cell diameter of 100 pixels and minimum size threshold of 30 pixels. Cell masks were expanded by 5 pixels and border-touching cells were excluded from analysis.
Morphometric measurements including area, perimeter, and eccentricity were calculated for both granules and cells using scikit-image v0.24 (Walt et al. 2014). All measurements were converted to physical units using the microscope’s calibrated pixel size (0.18 µm/pixel). For each cell, the number of contained granules was counted and normalized to cell area. The analysis pipeline was implemented in Python using cellpose, scikit-image, numpy, pandas and plotting libraries matplotlib, seaborn and microfilm v0.2.1.
Stringer, Carsen, and Marius Pachitariu. 2024. “Cellpose3: One-Click Image Restoration for Improved Cellular Segmentation.” February 12, 2024. https://doi.org/10.1101/2024.02.10.579780.
Stringer, Carsen, Tim Wang, Michalis Michaelos, and Marius Pachitariu. 2021. “Cellpose: A Generalist Algorithm for Cellular Segmentation.”Nature Methods 18 (1): 100–106. https://doi.org/10.1038/s41592-020-01018-x.
Walt, Stéfan van der, Johannes L. Schönberger, Juan Nunez-Iglesias, François Boulogne, Joshua D. Warner, Neil Yager, Emmanuelle Gouillart, and Tony Yu. 2014. “Scikit-Image: Image Processing in Python.”PeerJ 2 (June): e453. https://doi.org/10.7717/peerj.453.
Source Code
---title: Image Analysisjupyter: python3---Confocal microscopy images were analyzed using a custom Python pipeline. Maximum intensity Z-projections were generated for both DAPI and GFP channels. For granule detection, the GFP channel was preprocessed using Difference of Gaussians filtering (σ₁ = 3.0, σ₂ = 4.8) to enhance punctate structures while suppressing background noise. Granules were segmented using a global intensity threshold of 0.0009, followed by removal of objects smaller than 15 pixels and those touching image borders. Cell segmentation was performed using Cellpose v3.1 [@stringerCellposeGeneralistAlgorithm2021] with the cyto3 model [@stringerCellpose3OneclickImage2024]. Prior to segmentation, images were preprocessed with Gaussian smoothing (σ = 0.5 for GFP and σ = 1.0 for DAPI channels) and intensity rescaling to the [0,1] range. The model was configured with a target cell diameter of 100 pixels and minimum size threshold of 30 pixels. Cell masks were expanded by 5 pixels and border-touching cells were excluded from analysis.Morphometric measurements including area, perimeter, and eccentricity were calculated for both granules and cells using scikit-image v0.24 [@waltScikitimageImageProcessing2014]. All measurements were converted to physical units using the microscope's calibrated pixel size (0.18 µm/pixel). For each cell, the number of contained granules was counted and normalized to cell area. The analysis pipeline was implemented in Python using cellpose, scikit-image, numpy, pandas and plotting libraries matplotlib, seaborn and microfilm v0.2.1.```{python}# image analysisimport imageio.v3 as iiofrom cellpose import modelsfrom skimage.exposure import rescale_intensityfrom skimage.filters import gaussian, difference_of_gaussiansfrom skimage.morphology import remove_small_objectsfrom skimage.measure import label, regionprops, regionprops_tablefrom skimage.color import label2rgbfrom skimage.segmentation import clear_border, expand_labels# generalimport pandas as pdimport numpy as npimport refrom glob import globfrom os.path import basename# plot libsimport matplotlib.pyplot as pltimport seaborn as snsfrom microfilm import microplot``````{python}# pixel size = 0.18umPIXEL_SIZE =0.18IMAGES_PATH = ["data/ome-tiff/ddx3x_R326H/*.ome.tif", "data/ome-tiff/ddx3x_wt/*.ome.tif", "data/ome-tiff/ddx3x_L556S/*.ome.tif"]``````{python}def save_sample_images(fig_title, im_nuclei, im_cytoplasm, im_cytoplasm_filtered, im_granules_labeled, cells_labeled): fig, axs = plt.subplots(1,3, constrained_layout=True) im_cyto_g = gaussian(im_cytoplasm, 3) im_nuclei_g = gaussian(im_nuclei, 1) min_nuclei, max_nuclei = np.quantile(im_nuclei_g, [0.01, 0.999]) min_cyto, max_cyto = np.quantile(im_cyto_g, [0.2, 0.999]) microplot.microshow( images=[im_nuclei_g, im_cyto_g], cmaps=["pure_blue", "pure_green"], ax=axs[0], label_text=fig_title, label_font_size=5, unit='um', scalebar_unit_per_pix=0.18, scalebar_size_in_units=5, scalebar_font_size=10, scalebar_thickness=0.01, rescale_type='limits', limits=[[min_nuclei, max_nuclei], [min_cyto, max_cyto]]) microplot.microshow( images=[im_cytoplasm_filtered], cmaps=["magma"], ax=axs[1], rescale_type='limits', limits=[[0,0.005]]) axs[2].imshow(im_cytoplasm, cmap='gray') axs[2].imshow(label2rgb(cells_labeled), alpha=0.1)# axs[2].imshow(label2rgb(im_granules_labeled), alpha=0.1) axs[2].contour(cells_labeled, colors='white', linewidths=0.1) axs[2].contour(im_granules_labeled, colors='yellow', linewidths=0.1) axs[2].axis("off")# this line is specific to these data# fig_basename = re.search(r'\[(.*?)]', im_fn).group(1)# fig.suptitle(f'Image: {fig_basename}', fontsize=8) axs[0].set_title('Maximum projection', fontsize=6) axs[1].set_title('Channel after filtering', fontsize=6) axs[2].set_title('Detected cells and granules', fontsize=6) fig.savefig(f'figures/{fig_title}.png', bbox_inches ='tight', pad_inches =0, dpi=600)# fig.savefig(f'figures/{fig_basename}.pdf', bbox_inches = 'tight', pad_inches = 0, dpi=600)# fig.savefig(f'figures/{fig_basename}.svg', bbox_inches = 'tight', pad_inches = 0, dpi=600) plt.close(fig)``````{python}def get_treatment_from_image_filename(im_fn):if"ARS"in im_fn:return'ARS'elif'GLU-40min'in im_fn:return'GLU-40min'elif'CT'in im_fn:return'CT'else:raise(f"Fail in extract group from filename", im_fn)``````{python}def get_image_number_from_image_filename(im_fn): match = re.search(r'img(\d+)', im_fn) image_number = match.group(1) if match else'unknown'returnint(image_number)``````{python}def get_group_from_path(im_path):if'ddx3x_wt'in im_path:return'WT'elif'ddx3x_R326H'in im_path:return'R326H'elif'ddx3x_L556S'in im_path:return'L556S'``````{python}model = models.CellposeModel(gpu=True, model_type='cyto3')# Lists to store data for both dataframesgranule_data = []cell_data = []# plotfig_example, axs = plt.subplots(9,3, figsize=(9,28))example_number =3idx =0for im_path in IMAGES_PATH:for im_fn in glob(im_path):if get_treatment_from_image_filename(im_fn) =='GLU-20min':# skip 20min GLUcontinue#read image im = iio.imread(im_fn)# maximum intensity projection im_nuclei = im[0].max(axis=0) # nuclei is the first channel im_cytoplasm = im[1].max(axis=0) # cytoplasm is the second channel# Process cytoplasm and find granules im_cytoplasm_filtered = difference_of_gaussians(im_cytoplasm, 3) threshold =0.0009# we use a fixed global threshould because all images were collected at the same intensity im_granules = im_cytoplasm_filtered > threshold im_granules = remove_small_objects(im_granules, 15) im_granules_labeled = clear_border(label(im_granules))# Get granule properties granule_props = regionprops_table(im_granules_labeled, properties=['label', 'area', 'centroid', 'eccentricity', 'perimeter'])# match = re.search(r'imagem(\d+)', im_fn)# image_number = match.group(1) if match else 'unknown' granule_props['area'] = granule_props['area'] * PIXEL_SIZE * PIXEL_SIZE granule_props['perimeter'] = granule_props['perimeter'] * PIXEL_SIZE image_number = get_image_number_from_image_filename(im_fn) granule_props['image_number'] = image_number# granule_props['group'] = basename(im_fn).split(".lif")[0] image_treatment = get_treatment_from_image_filename(im_fn) granule_props['treatment'] = image_treatment image_group = get_group_from_path(im_path) granule_props['group']= image_group granule_data.append(pd.DataFrame(granule_props))# Segment cells with cellpose# preprocessing to improve cellpose segmentation im_cyto_pre = gaussian(im_cytoplasm, sigma=0.5) im_cyto_pre = rescale_intensity(im_cyto_pre,out_range=(0,1)) im_nuclei_pre = gaussian(im_nuclei, sigma=1) im_nuclei_pre = rescale_intensity(im_nuclei_pre,out_range=(0,1)) im_to_cellpose = np.stack([im_cyto_pre, im_nuclei_pre], axis=0) masks, flows, styles = model.eval(im_to_cellpose, channels=[0,1], diameter=100, min_size=30) cells_labeled = clear_border(expand_labels(masks, 5))# Process each cellfor cell in regionprops(cells_labeled): cell_mask = cells_labeled == cell.label granules_in_cell = im_granules_labeled[cell_mask] granule_count =len(np.unique(granules_in_cell[granules_in_cell >0])) cell_area = cell.area * PIXEL_SIZE * PIXEL_SIZE cell_data.append({'treatment': image_treatment,'group': image_group,'image_number': image_number,'cell_label': cell.label,'cell_area': cell_area,'granule_count': granule_count,'granules_per_area': granule_count / cell_area if cell_area >0else0 }) fig_title =f"DDX3X-{image_group}-{image_treatment}-img{image_number}"if image_number == example_number:# plot one image from each group im_cyto_g = gaussian(im_cytoplasm, 3) im_nuclei_g = gaussian(im_nuclei, 1) min_nuclei, max_nuclei = np.quantile(im_nuclei_g, [0.01, 0.999]) min_cyto, max_cyto = np.quantile(im_cyto_g, [0.2, 0.999]) microplot.microshow( images=[im_nuclei_g, im_cyto_g], cmaps=["pure_blue", "pure_green"], ax=axs[idx, 0], label_text=fig_title, label_font_size=5, unit='um', scalebar_unit_per_pix=0.18, scalebar_size_in_units=5, scalebar_font_size=10, scalebar_thickness=0.01, rescale_type='limits', limits=[[min_nuclei, max_nuclei], [min_cyto, max_cyto]]) microplot.microshow( images=[im_cytoplasm_filtered], cmaps=["magma"], ax=axs[idx,1], rescale_type='limits', limits=[[0,0.005]]) axs[idx,2].imshow(im_cytoplasm, cmap="gray") axs[idx,2].imshow(label2rgb(cells_labeled), alpha=0.1) axs[idx,2].contour(cells_labeled, colors='white', linewidths=0.1) axs[idx,2].contour(im_granules_labeled, colors='yellow', linewidths=0.1) axs[idx,2].axis("off")# names and labelsif idx ==0: axs[idx, 0].set_title('Maximum projection', fontsize=8) axs[idx, 1].set_title('Channel after filtering', fontsize=8) axs[idx, 2].set_title('Detected cells and granules', fontsize=8) idx +=1# save all images to verify the quality save_sample_images(fig_title, im_nuclei, im_cytoplasm, im_cytoplasm_filtered, im_granules_labeled, cells_labeled)# Create final dataframesgranules_df = pd.concat(granule_data, ignore_index=True)cells_df = pd.DataFrame(cell_data)# Save figure with examplesfig_example.savefig('figures/examples.png', bbox_inches ='tight', pad_inches =0, dpi=600)fig_example.savefig('figures/example.pdf', bbox_inches ='tight', pad_inches =0, dpi=600)fig_example.savefig('figures/example.svg', bbox_inches ='tight', pad_inches =0, dpi=600)``````{python}cells_df.to_csv("output/granules_per_cell.csv", index=False)granules_df.to_csv("output/granule_area.csv", index=False)```